

Archived - Use of Data from the Electronic Health Record for Health Research – current governance challenges and potential approaches
This page has been archived on the Web
Information identified as archived is provided for reference, research or recordkeeping purposes. It is not subject to the Government of Canada Web Standards and has not been altered or updated since it was archived. Please contact us to request a format other than those available.
Donald J. Willison Sc.D.
Associate Professor, Dept. of Clinical Epidemiology & Biostatistics, McMaster University
Commissioned by the Office of the Privacy Commissioner of Canada
March 2009
Disclaimer: The opinions expressed in this report are those of the author(s) and do not necessarily reflect those of the Office of the Privacy Commissioner of Canada.
Acknowledgements:
This paper was commissioned by the Office of the Privacy Commissioner of Canada. The views and opinions contained in this document are those of the author and do not necessarily reflect the views and opinions of the Office of the Privacy Commissioner of Canada, or of the Government of Canada.
I would like to thank Patricia Kosseim, Philippa Lawson, and Ed Brown for their helpful comments to an earlier draft of this paper. My thoughts in this area have also greatly benefitted from numerous discussions with Elaine Gibson and Kim McGrail over another paper being written concurrently. Thanks, also, to Sue Johnston and Jennifer Ranford for their editorial feedback.
Executive Summary
Academic researchers look, in anticipation, toward the possibilities that the common interoperable electronic health record (EHR) will offer enhanced ability to conduct a wide variety of health-related research. At the same time, access to data from a system-wide interoperable EHR poses a number of challenges that have, to date, not been addressed. Canada Health Infoway acknowledges that there will be demands for secondary use of the EHR for research and raises critical questions about: whether and how people should be informed of those uses; what level of de-identification is required before this information may be used ethically for research without requiring consent; and whether the EHR should record whether a patient agrees to be contacted for health research. It is the purpose of this paper to examine current challenges in governing research use of health information and potential approaches to addressing these issues in the context of the common interoperable EHR.
This paper has three sections. Section 1 is, for the most part, a primer for the reader who is unfamiliar with the range of potential research uses of health information – whether from the EHR or from the many other potential sources. Most of the research uses discussed are observational in nature: safety and effectiveness research; public health; occupational health and safety; quality improvement research; health policy and health services research; population health; and translational bioinformatics. The reader who is familiar with these uses may skip to Section 2.
Section 2 identifies a number of inter-related challenges in the governance of research use of health information generally, and some novel challenges in the context of the interoperable EHR including:
- The richness of the linkable person-level data required for most research makes it virtually impossible to sufficiently de-identify the data to the point where they are, in effect, anonymous.
- The indistinct boundaries between research and a number of other secondary uses of the data that make for wide disparities in the ethical oversight (including privacy protection) between research and these other secondary uses.
- The growing concern that current ethical standards around consent for use of personal information for health research and the current two-stage consent process are introducing substantial biases into research results and substantial additional costs.
- The shift in how research is conducted. Increasingly, teams of researchers are developing data repositories (registries) and biobanks that serve as platforms for a wide variety of future research which challenge existing Fair Information Principles around limits on data collection, use, and retention. There is currently no systematic documentation of the existence of these registries and biobanks.
- The poor fit between conventional consent models for research and the development of research registries and biobanks that will serve as platforms for a broad array of future research uses. .
- The absence of effective mechanisms for eliciting consent choices for the wide range of potential research uses of one’s personal health information. The time and costs involved may argue for the impracticability of obtaining consent at all in the current environment. However, advances in the interoperable EHR – in particular, the inclusion of a patient portal into their health records – offer potential solutions.
- Overlapping jurisdictions in the oversight of overall research uses of health information that could lead to a vacuum in leadership in governance.
These challenges and corresponding recommendations are summarized in Tables 1 and 2 that immediately follow the executive summary.
The challenges posed in Section 2 call for a fundamental re-consideration of how information is used for research and the role of research vis-à-vis clinical care, public health, and quality improvement. Section 3 picks up this challenge by exploring what a coherent information management system might look like. Section 3.1 calls for the dissolution of the artificial boundaries between research and quality improvement, system planning, public health, and the other secondary uses that generally receive little ethical scrutiny. It proposes a proportionate approach to ethical review of all secondary uses, depending on the level of risk posed to those whose information is being used. It also calls for a re-conceptualization of the primary uses of health information to include the management of the health of individuals and populations, in addition to management of the health care system, including research that supports these uses. This still leaves open the issue of the amount of control that individuals should have on the research use of that information.
Section 3.2 suggests that new approaches to research use of personal information in long-term research repositories calls for an expanded approach to consent and recognition that different research uses may warrant different default consent choices. Here, suggestions are made for ways of eliciting and documenting consent choices, and for simplifying the choice process to avoid decision overload. One promising future approach – the use of a patient portal into one’s health record – offers particular promise. It would provide opportunity for individuals to document and communicate their consent choices and to be informed of the uses of their personal information. A broader range of consent options than the conventional full consent or exemption from consent is also suggested and explained. One option is to assign different default consent options for different types of research, with some ability (in most cases) of individuals to modify the default option. These default consent options would need to be determined through a consensus development process involving researchers, ethicists, regulators, and the public.
To maintain public trust in the use of their personal health information, access to data for research will require strict safeguards. Decisions need to be made regarding who may develop research repositories and under what conditions. Section 3.3 outlines options for access and disclosure control and for governance over the review of individual research projects, the development of research repositories and institutions’ information use practices.
To improve consistency in decision-making across research ethics boards regarding the conditions under which researchers may use health information, it would be useful to develop a set of case studies that represent common types of observational research. These should contain sufficient detail and analytic commentary as to permit the identification of critical factors that affect decisions as to the role of consent, necessary safeguards, and limitations on access and disclosure. In the case of research involving the common interoperable EHR and biological samples, consideration should be given to developing centralized specialty review bodies that have the capacity to review research protocols involving access to data from the EHR and use of biological samples.
The interoperable EHR could make it much easier for the proliferation of a range of databases. This section suggests managing the proliferation through regulation, requiring the approval and registration of all research registries, databases, or biobanks, subject to a range of strict guidelines.
Given the current leadership vacuum in research governance at the overall systems level, the federal and provincial privacy commissioners/ombudsmen should consider whether they wish to take a lead in this regard. If so, actions should be done in consultation with the other potential governors of these research uses of health information. Whichever body takes ultimate responsibility for this function, one approach is to develop a reporting structure such that institutions maintain an inventory of:
- The databases and biobanks within their organization;
- The research studies that emanated from the databases;
- Any disclosures of the data to other institutions.
The research data repositories will become potent analytic tools. Once established, there will be need for effective mechanisms to limit their uses to the original research purposes.
Finally, Section 3.4 suggests ways in which the many affected parties – but particularly the public – could be engaged in this process. Initially, this could consist of consultations that focus on obtaining feedback on the acceptability of proposed models and conditions of research use. There would also be need for a concerted educational phase, to raise public awareness of the various applications of their health information beyond direct patient care, and the options and mechanisms available to them to control the use of their information. Serious consideration should be given to designing meaningful and substantive public involvement in the ongoing governance process that is developed. This would assist with issues such as commercialization of research outputs.
In conclusion, this paper frames both observational health research and privacy protection as two public goods. Areas of tension exist between the two, and suggestions are made as to how they may successfully co-exist.
Changes at the margin are not sufficient. Attention needs to be given sooner rather than later to the matter of necessary governance and infrastructures, lest the current process lead to further insufficient and inefficient ad hoc amendments to the governance over research rather than the establishment of a holistic solution.
| Section | Recommendation | Relevant Fair Information Principle* | |||||
|---|---|---|---|---|---|---|---|
| Identifying purpose | Consent | Limiting collection | Limiting use, disclosure, & retention | Safeguards & Governance | |||
| General Challenges: | |||||||
|
1. Most health research requires linkable person-level data. Privacy laws address use of personal information – i.e. information that can identify individuals either directly or indirectly, through reasonably foreseeable means. Health researchers generally require linkable person-level data for their analyses. In most cases, the identity of the individual is immaterial to the analysis. Even if direct identifiers (name, address, social insurance number, etc.) are removed, the remaining records are sufficiently rich in indirectly identifying information that they must be managed as if they were identifiable (i.e. personal). Those data elements that are particularly “risky” for re-identification are also of particular interest to researchers. For example:
This raises both technical and procedural challenges in the disclosure of the data for research. These challenges are not insurmountable but require a level of expertise in de-identification that is currently not accessible to most data custodians. |
2.1 | 9 – 11 | |||||
|
2. Indistinct boundaries. The boundaries between research and a number of other well-recognized secondary uses of the data are indistinct. For example:
Yet, there are major differences in the degree of ethical oversight between research and these administrative secondary uses. If labelled “research” the activity receives ethics review, including consideration of whether individual consent is required. If not labelled research, this activity often receives no ethical scrutiny whatsoever and individuals are not generally notified or given the opportunity to opt-out. There is also an increasing blur of research and clinical care – particularly in the areas of genomics and post-market evaluation of drugs and devices. |
2.2 | 1 – 3 | |||||
|
3. Changing nature of research. Increasingly, single-purpose research projects with a limited number of investigators are giving way to the development of data repositories (registries) and biobanks that will serve as platforms for a wide variety of future research of much larger teams of researchers. Because it is not possible to anticipate all the research questions at the time of developing the research repository, it is not possible to satisfy the Fair Information Principles around limits on data collection. Essentially, all the data in the health record could be relevant to future research questions. This also requires a broader interpretation of the Fair Information Principle limiting data use and retention to be compatible with the broader intended use of the data. |
2.3 | 4 – 7, 14 | √ | √ | |||
| Challenges related to consent | |||||||
|
4. Poor fit of current consent models with observational research. Conventional consent models for research fit poorly with the use of large datasets in an epidemiologic context. Current consent models tend to be driven by the requirements of clinical trials. The dichotomous approach of full project-specific consent vs. exemption from requiring consent falls short of the broader range of consent possibilities in the context of observational use of data across a spectrum of research. |
2.4.3 | 4, 5 | √ | ||||
|
5. Lack of effective mechanisms for eliciting consent choices for the wide range of potential research uses of one’s personal health information. In the absence of such mechanisms, the time and costs involved will provide substantial grounds for arguing the impracticability of obtaining consent. |
2.4.3 | 6 – 8, 17 | √ | ||||
|
6. Impact of current consent requirements on research. There is growing concern that current ethical standards regarding consent for use of personal health information have a negative impact on research through the introduction of selection bias and increasing the cost of conducting research. This applies to both:
|
2.4.2 | 7 | √ | ||||
|
7. Lock-box provisions. It is unclear whether lock-box provisions apply to research use of de-identified data where there will be no attempt to contact the individual. |
2.4.5 | 6 – 8 | √ | ||||
|
8. Commercialization of research. The move toward commercialization of research discoveries creates tensions, as research indicates people desire greater control over research use of their data when there is a commercial element. |
2.5.3 | 6 – 8 | √ | ||||
| Challenges related to limiting use, disclosure, and retention | |||||||
|
9. Initiatives to improve access to research data. International research initiatives by public research granting agencies to make raw data more readily available to other researchers exist in tension with the Fair Information Principle limiting disclosure of data. These initiatives are largely motivated by a desire for better stewardship of resources by making greater use of the investment made in the initial collection. |
2.5.1 | 14 | √ | ||||
|
10. Data retention. Researchers are reticent to destroy data out of concern for the difficulty and cost in re-constructing the dataset, if required, for re-analysis of data or audit trails, in the event research results are challenged. |
2.5.2 | 14 | √ | ||||
|
11. Potential loss of raw data for research. The Fair Information Principle limiting data retention may result in the loss of critical data documenting exposure to health hazards if there is a latency period of decades before a particular illness is manifest. |
2.7 | – | √ | ||||
| Governance-related challenges | |||||||
|
12. There is currently no systematic inventory of the existence of registries and biobanks. Nor are there common criteria as to who may develop and manage these research repositories and under what conditions. While the large data institutes have high standards for secure management of data in their possession, smaller research units pay uneven attention to these issues. |
2.6.2 | 13, 14 | √ | ||||
|
13. Variation in REB decisions. There is considerable variation in REB decisions on when research may be exempted from requiring consent. It is not clear that differences across REBs for the same protocol are justifiable. |
2.4.1 | 11, 12 | √ | √ | |||
|
14. Overlapping jurisdictions in the oversight of overall research uses of health information runs the risk of a vacuum in leadership in governance. |
2.6.3 | 15, 19 | √ | ||||
|
15. Need to secure the public’s trust in the research use of personal health information. |
16–18 | ||||||
|
16. Lack of consensus among affected parties. An ongoing challenge is that the number of parties with a voice in research use of PHI is large – researchers, ethicists and lawyers, physicians and other data custodians, governments, patients, and family members. This slows progress in effecting change, particularly if interactions promote entrenchment in positions. |
16 | ||||||
| * The Fair Information Principles are a set of procedural guidelines for the collection, management, processing, and safeguarding of personal information. In 1981, the member countries of the Organization for Economic Cooperation and Development agreed to these principles, which are now are at the core of most of the privacy legislation in the Western world. In Canada, the Canadian Standards Association re-framed the Fair Information Principles in its Model Code for the Protection of Personal Information to include 10 principles: accountability; identifying purpose; obtaining consent; limiting collection; limiting use, disclosure, and retention; ensuring accuracy; safeguards; openness about information use policies; allowing individuals access to information about them; and allowing people to challenge compliance with the Principles. (Canadian Standards Association, 1996) For the most part, the Fair Information Principles are consistent with good research practices. This table identifies the five principles where there are challenges with current and evolving research practices. | |||||||
| Section | Challenge | Planning horizon | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Short | Medium | Long | ||||||||
| Re-thinking the role of research vis-à-vis other secondary uses of personal health information | ||||||||||
|
1. The artificial boundaries between research and practice in the areas of quality improvement, systems planning, and public health should be dissolved. Research uses of personal information should be treated equally to these other secondary uses and ethical review should be applied to all secondary uses, proportionate to the risks associated with these data uses. |
3.1 | 2 | √ | |||||||
|
2. Practical frameworks need to be developed for evaluating risks and benefits of these secondary uses of health information. |
3.1 | 2 | √ | |||||||
|
3. In the long run, consider re-conceptualizing the primary uses of personal health information to include the management of the health of: individuals, populations, and the health care system. Research supporting these activities would also constitute primary uses. Such a shift would call for open public deliberation. |
3.1 | 2 | √ | |||||||
| Consent | ||||||||||
|
4. It has been suggested that we move from individual consent for particular acts of collecting, using, or storing data to a public mandate for systems within which data are collected, stored, used, and disclosed. (O'Neill, 2001) Such a move will require public deliberation and consensus development. |
3.2.1 | 3, 4 | √ | |||||||
|
5. This public mandate does not obviate individual choice. At the individual level, we should consider a more expansive approach to consent that includes broad opt-in to a program of research and notification with opt-out. |
3.2.2 | 3, 4 | √ | |||||||
|
6. Mechanisms need to be developed for both eliciting and documenting in advance individual consent choices for different types of research uses of one’s personal information. Suggestions for this are offered in the text. The same process could be used to determine people’s preferences for how to be approached for participation in research studies. |
3.2.3 | 3 – 5 | √ | |||||||
|
7. To minimize decision overload, default consent choices are suggested for different types of research uses. Default settings should reflect: (a) the societal benefit of the research vs. the risks to the individual; and (b) the potential for individual profit. For example, recognizing their broader societal benefits, in the cases of public health and post-marketing surveillance of drugs and devices (whether or not formally defined as “research”), the default may be mandatory inclusion of one’s data. For quality improvement and population health research, the default may be notification with opt-out. For studies involving biological samples, the default may be a broad opt-in. (See Table 1 for details.) Again, the default consent choices should be determined through a public deliberative process. |
3.2.3 | 5 – 8 | √ | |||||||
|
8. Research is required into logistical challenges, including: equity in access to mechanisms for expressing one’s consent choices (e.g. through patient portals to their EHR); how to manage expectations of just how much control will be offered through the patient portal; how to ensure that the opting-out process is not done capriciously; and the impact of commercialization of products of innovation on consent choices. |
3.2.3 | 5, 7 | √ | |||||||
| Context within which research would be conducted | ||||||||||
|
9. The practice of directly posting raw data online – even if de-identified – should be discouraged. Instead, the existence of these repositories could be posted online with mechanisms to ensure the data are readily available to researchers for bona fide research purpose, with appropriate disclosure controls and user agreements in place. |
3.3.1 | 1, 9 | √ | |||||||
|
10. Secure data repositories should continue to be developed, with appropriate access and disclosure control mechanisms in place. New technologies that enable secure remote access to these repositories should be adopted and both data custodians and researchers should be trained in their use. |
3.3.1 | 1 | √ | |||||||
|
11. To improve the consistency in the decisions of REBs regarding the conditions for exemption from requiring consent for research, detailed case studies should be developed and deployed as part of a continuing education outreach to research ethics boards and researchers. These case studies could build on the earlier case study work by the CIHR in 2002. |
3.3.2 | 13 | √ | |||||||
|
12. In the case of research involving the common interoperable EHR and biological samples, consider developing centralized specialty review bodies that have the capacity to review research protocols involving access to data from the EHR and use of biological samples. |
3.3.2 | 13 | √ | |||||||
|
13. Data and tissue holdings intended for research purposes should be registered with the research ethics boards of the institutions where they are housed. Any research projects that emanate from these holdings should also be collated in this database. With the registration of databases and biobanks, it would then also be possible to keep track of particular studies that have been conducted using these data sources. |
3.3.2 | 12 | √ | |||||||
|
14. Wherever possible, data and sample repositories should be under the stewardship of institutions rather than individual researchers. In this way, long-term secure management of research data may more readily be assured. With appropriate access rules, this would also permit the repositories to be unrestricted in data available while better satisfying the Fair Information Principle of limiting use and retention for individual research projects. |
3.3.2 | 3, 9, 10, 12 | √ | |||||||
|
15. Given the current leadership vacuum in governance at the overall systems level, the federal and provincial privacy commissioners/ombudsmen should consider whether they wish to take a lead in this regard. If so, actions should be done in consultation with the other potential governors of these research uses of health information. |
3.3.2 | 14 | √ | |||||||
|
16. Operational models of use of personal health information for research (infrastructures, consent approaches, governance) should be vetted with all affected parties, including the public, in a variety of forums. Initial efforts should create opportunities for inviting feedback on the acceptability of proposed models of research use. |
3.4 | 5, 15 | √ | |||||||
|
17. Ongoing efforts will be needed to raise public awareness of what uses are made of their personal information for health research and what options are available for individuals to control the use of that information. |
3.4 | 15 | √ | √ | ||||||
|
18. Design meaningful ongoing lay input in the overall governance structure. |
3.4 | 14, 15 | √ | √ | ||||||
1. Background
1.1 Context and purpose of this paper
In the health care field, the transfer from paper to electronic records has been a relatively slow process that has proceeded in a patchwork fashion. Since 2001, efforts have been underway, through Canada Health Infoway, to create a pan-Canadian interoperable electronic health record (EHR). While Infoway acknowledges that there will be demands for secondary use of the EHR for research, it has taken an incremental approach to the roll-out of the design architecture and currently regards secondary research use as beyond its purview. (Anonymous, 2007b)
The secondary research uses of health information generally are of great interest to the Office of the Privacy Commissioner of Canada. The introduction of a pan-Canadian EHR raises important questions as to: whether and how people should be informed of those uses; when consent may be required and when research may be exempted from requiring consent; how one’s willingness to participate in research might be recorded in the EHR; and the governance over use of information from the EHR for research purposes. These issues are best considered and addressed in advance of such uses. The purpose of this paper is to address these questions.
This report describes current and anticipated research uses of personal health information (PHI) from the EHR, the privacy-related issues and challenges that need to be addressed as information from the EHR is accessed for research, and potential approaches to addressing these issues and challenges. The report focuses on observational research in which there is no active intervention. There are generally two purposes for researchers to access information in the health record: (1) to extract existing data from the record and possibly link the data with records from other sources, with no interest in the identity of the individual and no intention of direct contact with those individuals; (2) to contact patients – for example, to invite participation in a study which requires direct data collection from that individual. In this case, they usually request that certain information from the health record be gleaned so as to target the focus of their recruitment efforts. The data may be collected through a survey or questionnaire. In addition, there may be physical measures taken, such as blood pressure, and urine or blood samples may be taken for laboratory analysis.
This report is divided into three sections. Section 1 provides background information for the reader unfamiliar with the various types of health research that may use information from the health record. Although the focus is not on clinical trials, they are briefly described as clinical trials share the need for access to existing health information to screen for potential research participants, and they provide a reference point for some of the observational research methods, such as post-marketing surveillance. Section 2 describes several inter-linked ethical challenges that sometimes go beyond a strict consideration of privacy, but link with privacy concerns. Section 3 proposes approaches to addressing these challenges.
Themes that emerge throughout this report include:
- The indistinct boundaries between research and public health, quality improvement, and even clinical care;
- Challenges with conventional approaches to consent, including
- satisfying the disclosure requirements in contemporary observational research
- mechanisms to support patient choice regarding research use of their PHI; and
- The physical infrastructures, human resources, and governance mechanisms that need to be in place to manage research uses of PHI in a secure fashion.
1.2 Health research applications of information from the EHR
Existing data sources have long been used to answer clinical and epidemiologic research questions in a timely and efficient fashion. Studies that would involve years or even decades to conduct through prospective gathering of data may be accomplished in months. The expectation of researchers is that the advent of the common interoperable EHR will increase further the speed and efficiency of research, and enable the answering of a much greater breadth of health-related questions.
Currently, most of the types of research described in this section use personal information from a variety of data sources, including conventional paper health records, registriesFootnote 1 and administrative databases that are chiefly used for claims adjudication. In addition, health researchers may access information on non-medical determinants of health, such as education and income. This section describes the range of types of health research that use personal information and, as appropriate, how these may be affected by the introduction of the interoperable EHR.
1.2.1 Clinical trials:
Clinical trials (also known as randomized controlled trials, randomized clinical trials, or RCTs) are studies of interventions to ascertain the efficacyFootnote 2 of specific therapies, procedures, or devices in a very specific target patient population. The chief secondary uses of existing health records are for identifying health care facilities that have a high-enough volume of patients of interest that they could serve as recruitment sites and potential research participants whom they may approach to participate in clinical trials.
Where the health condition of interest is a chronic illness, potential research participants may be identified through medical record review. When using a paper record system, records are initially screened drawing from a substantially larger pool of potential research participants than the final list of eligible patients. It is then necessary for an individual to glean through multiple sections of the records of this larger pool of individuals to determine who qualifies for inclusion in the RCT. The screener is often a research assistant. There has been discussion in bioethics circles as to who should be permitted to scan the health records to identify potential research participants, and how these eligible individuals are subsequently approached to participate. In general, the preferred practice is that this be conducted by someone with reasonable expectation of access to that information for care or administrative purposes. Exceptions may be made due to limited staffing. An appropriately designed electronic health record system could be less privacy invasive to the extent that it would reduce the number of charts of ineligible patients initially screened manually by an individual not involved in the circle of care, and by providing an automated screening process on one or more diagnostic fields.Footnote 3
1.2.2 Safety and effectiveness studies
While considered the “gold standard” method for assessing efficacy of new treatments, clinical trials have shortcomings for measuring safety and real-world effectiveness of new therapies. In particular:
- Sample sizes are usually too small to detect all but the most common adverse effects of the therapy. In some cases, it is not until a treatment is used in thousands of patients that less common (or less distinctive) adverse events may be detected.
- While providing information about the efficacy of the treatment under ideal circumstances, most trials do not provide adequate information about the effectiveness of the treatment (i.e. how well it works under real-world conditions in which patients have concurrent illnesses and have less-than-perfect compliance with medication taking when compared with current best practice as opposed to placebo).
Currently, much of the information for adverse event monitoring comes from spontaneous reports that may serve as “signal generators” to alert researchers as to the potential of a specific adverse event associated with a therapy. If there is suspicion of an association between exposure to a certain treatment and some adverse health event, it is often tested by conducting a retrospective review of existing data, combining two or more administrative and/or clinical records. For example, an association between exposure to benzodiazepinesFootnote 4 and fractures due to falls in seniors was determined through linking drug reimbursement datasets with hospitalization discharge abstracts, using a common patient identifier. (Monane & Avorn, 1996) These record-linkage studies can be slow and laborious.
Researchers would likely turn to the common interoperable EHR system as the preferred data source for such research, as the clinical dataset will permit much higher fidelity analyses than are possible with administrative datasets. Researcher access to an interoperable EHR system may also promote more data mining studies where associations are tested between exposures to different treatments and a wide range of health outcomes, without prior hypothesis of a causal relation with a particular outcome (i.e. hypothesis generation).
A variety of systematic approaches to post-marketing data collection for evaluating safety, effectiveness, and efficiency is being contemplated under a federal initiative for progressive licensingFootnote 5 of pharmaceuticals in Canada. One American policy that has captured interest in Canada is called “coverage with evidence development”. (Pearson, Miller & Emanuel, 2006) In these circumstances, patients wishing a particular marketed therapy must either permit ongoing monitoring of their personal health information or they must enrol in a pragmatic randomized clinical trialFootnote 6 in order to receive the treatment at no charge. It is currently unclear whether individuals would have the ability to opt-out of such a monitoring scheme without having to pay for the therapy. In the presence of an interoperable EHR, data collection may be accomplished, in part, through periodic automated downloading of data from various data repositories in the common EHR to some central research site. It may also involve additional data collection that would not be usual and customary outside a research environment. This increases the blur between what is research and what constitutes the fulfilling of one’s obligation to review new drugs and devices for adverse events as part of a progressive licensing scheme.
1.2.3 Public health / occupational health and safety
Public health takes a broad approach to what determines the health of populations, including the food chain, our water supply, and environmental contaminants. As such, it draws upon a much broader range of data than simply health information.
There are two main ways in which health information is used in public health:
- Routine surveillance, particularly:
- Communicable diseases (e.g. influenza, STDs, MRSA)
- Chronic illnesses (e.g. diabetes)
- Ad hoc analyses. For example:
- adverse health events associated with exposure to environmental toxins, such as waste dumps, workplace chemicals, high-voltage power lines
- follow-up on food-borne illness outbreaks
For some communicable diseases, reporting of cases is mandatory. This form of monitoring tends to focus on geographic distribution of events, which raises risk of re-identification. With the advent of the common interoperable EHR, gleaning of data for public health reporting is likely to become automated. In many cases, the distinction between routine public health monitoring and public health research using the same data is unclear.
The ad hoc public analyses are even more likely to blur with research activities. The methods employed to study these associations are virtually identical. There is a high likelihood that the report will be published in a peer-reviewed journal. Data sources for these studies depend upon the research question at hand and may include primary data collection through surveys, gleaning of data from hospital discharge abstracts, or even chart review.
1.2.4 Population health
Like public health, population health research takes a much more expansive view of the determinants of health to include, for example, income, education, housing and environmental factors. These studies usually involve analysis of large and diverse datasets, combining information on health and social services utilization, school records, income, and socio-spatial data. Because of the complexity of organizing these data and the potential for both use and misuse of the data, efforts are underway to establish centralized data centres that will serve as platforms for a variety of research. The intention is that these data enclaves will provide both the expertise at linkage and interpretation of the data, and high levels of data protection.Footnote 7
1.2.5 Quality improvement research
As with public health, it is increasingly difficult to distinguish between the practice of quality improvement and research in quality improvement. The methods employed are very similar. Both Q.I. research and routine Q.I. are heavily dependent on retrospective medical record review, although either may involve prospective studies that collect additional data. Q.I. research is very heterogeneous. The use of data from the EHR will vary greatly. Common elements include screening specific diagnostic, intervention (drugs, surgery, devices) or outcome fields to assemble cohorts of patients of interest and test of associations between various interventions and outcomes.
1.2.6 Health policy / health services research
Canada’s provincially-administered health care system has served as a natural laboratory for examination of the impact of health policies on health care provision and on health outcomes. Use of health information for these comparisons involves multi-jurisdictional comparisons of policies and outcomes over time – both within province and particularly among provinces. To date, inter-provincial comparisons have been ad hoc, usually involving exporting of data from one site to another. Health services and policy research is currently heavily encumbered by challenges with assembling individual-level comparative data across provinces. The challenges involved are political, legal (differences in privacy legislation across provinces), and definitional (i.e. the lack of standardized definitions or documentation of essentially identical services or actions). The common EHR would resolve some of the technical problems of data access, but policies and procedures would need to be developed for determining how comparative analyses would be conducted, for example, through:
- Centralized pooling of data into a common data warehouse and refining to create common datasets; or
- Parallel data warehouse structures across provinces with capacity to conduct parallel analyses.
1.2.7 Translational bio-informatics
Translational bioinformaticsFootnote 8 is a relatively new area of research. It attempts to draw association between genetic variations and health. Currently most research activities focus on testing specific hypotheses regarding the association between variants in small segments of DNA (known as SNPs) and specific health manifestations (e.g. obesity, hypertension, susceptibility to a particular form of cancer). The goal, however, is to draw broad associations – both positive and negative – between variations in the human genome with a wide array of expressed health conditions. Ultimately, this type of research will involve mapping individuals’ entire genome and linking this information with information from the electronic health record, with a view to conducting large-scale data mining across hundreds of thousands of individuals. By its nature, data mining does not test specific hypotheses as to causal associations between a particular gene expression and a health condition. Instead, it identifies associations that may be tested in the future. Therefore, no specific research question is identified.
Whole genome mapping is currently impracticable on a large scale, largely due to capacity and cost. Currently, the cost of mapping a single whole genome (approximately 3 billion base pairs) is in the range of $100,000. The National Institutes of Health have set the goal of reducing the cost of whole genome sequencing to $1000 by 2014, so as to make it economically feasible to include in routine medical care. (Anonymous, 2008c) It is anticipated that this will be technically feasible before that time. (Shaffer, 2007)
There are also major technical challenges associated with automatically gleaning data from the EHR. Much of the information in the EHR will be entered in free-text format. Therefore, there is considerable research to develop algorithms to permit the extraction of target data from free text fields to provide reliable and valid markers of the presence of particular diseases. (Meystre, Savova, Kipper-Schuler & Hurdle, 2008) There are also efforts underway to determine ways of encrypting the information in the record, so as to anonymize the data. (Adida & Kohane, 2006)
Currently, there is a handful of sites in the USA conducting large-scale record linkage between the genome and the EHR. Most of these sites sufficiently de-identify the data so as to be exempted under existing U.S. regulations from requiring consent for the use of these samples, which are often derived from surplus tissue collected in the course of treatment. However, it has recently been shown that it is possible to re-identify these sequenced DNA data that are legally considered anonymous. (Couzin, 2008; Malin & Sweeney, 2001; McGuire & Gibbs, 2006)Footnote 9 How this will play out in the Canadian context remains to be seen, but this is an area requiring attention, as efforts are underway in several jurisdictions to launch bioinformatics initiatives in Canada. Ethical considerations around consent, re-contacting, and governance over the research activities are likely to be heavily influenced by the recent international consensus statement that includes very prominent Canadian and American bioethics and legal scholars. (Caulfield, McGuire, Cho, Buchanan, Burgess, Danilczyk et al. 2008)
2. Issues and Challenges
2.1 Most health research requires linkable person-level data
For the majority of the research described above, researchers have no interest in the actual identities of individuals.Footnote 10 However, individual-level linkable data are required for several reasons:
- For analytic purposes. When studying the relationship between some exposure (a drug, a procedure, a policy) and a health outcome, individual-level data allows the researcher to obtain a more precise estimate of the relationship between exposure and outcome by “controlling” for other factors that have an impact;
- To look at spill-over consequences of policies. Attempts to limit expenditures in one budget portfolio (e.g. pharmaceuticals) may result in unanticipated increase in utilization in other portfolios (e.g. physician visits, emergency department visits). Therefore, any evaluation of policies should examine spill-over effects to other budget envelopes. This is often referred to as “squeezing the balloon”. To address this, it is usually necessary to combine files from disparate data sources, including non-medical determinants of health. In the current non-integrated data environment, one needs a common identifier to most effectively link records to examine this.
- For cohort studies that follow particular patient groups prospectively, it is necessary to update files from time to time. A common identifier enables this.
In most cases, direct identifiers may be readily removed without affecting the research, so long as it remains possible to link disparate health records, as the identity of individuals is not material to the research. However, the remaining data are often still sufficiently rich in indirectly identifying variables (e.g. date of birth, geographic locators, dates of admission, discharge, specific procedures) as to render the data re-identifiable through indirect means.
There is a considerable body of literature addressing ways of minimizing the risk of re-identification through statistical disclosure control methods. (Anonymous, 1994; El Emam, Jonker, Sams, Neri, Neisa, Gao et al. 2007; Eurostat, 1996) Many of the methods are quite technical and currently require an expertise that is not widely available. Decisions as to the most appropriate technique to use and variables to mask requires several judgment calls, based on the specific questions being asked in the research protocol. Therefore, for all practical purposes, even data with direct identifiers removed should be managed with a view to their potential for re-identification.
2.2 Indistinct boundaries between research and other secondary uses, and even with clinical care
Reference is commonly made to primary and secondary uses of information from the health record. Generally, primary uses are those related to the management of the care of an individual patient. The term “secondary uses” has been given to all other uses. Under the Fair Information PrinciplesFootnote 11 that form the basis of most modern data protection legislation in the Western world, unless impracticable, any secondary uses of information for research should be subject to individual consent.
Historically, a number of secondary uses of information from the health record – such as quality improvement, risk management, health systems improvement, infection control, and public health – have been permitted without the requirement for any additional consent. Little is known about health care institutions’ notification policies regarding secondary uses of personal health information in their custody and, for these secondary purposes, patients are generally not given the opportunity to opt-out.
Secondary use of the same personal information for health research is treated quite differently. Generally, research uses of health information are not permitted without explicit consent, unless an exemption is granted by a research ethics board. However, as noted above, there is considerable overlap in the boundaries between research and quality improvement, public health, and systems management. From a policy perspective, this is a substantial challenge. (Kosseim & Brady, 2008) For example, if the secondary use were deemed “quality improvement”, there is an exemption from requiring individual consent and very little scrutiny over use of that information. If that use were deemed “research”, individual consent may be required. In addition, there will be reviews by one or more academic research ethics boards and data custodians. At the same time, there is growing concern that current consent requirements and the review process are constraining researchers’ ability to conduct observational and epidemiologic research.
The distinction between research and clinical care also starts to become less distinct. For example, ongoing post-marketing evaluation of drugs and devices through pragmatic trials and programs such as coverage with evidence development are introducing research into everyday clinical practice, blurring the treatment relationship between physician and patient. (Fransen, van Marrewijk, Mujakovic, Muris, Laheij, Numans et al. 2007; Maclure, Carleton & Schneeweiss, 2007; Miller & Pearson, 2008; Pearson, Miller & Emanuel, 2006)
Further, in the areas of genomics and translational bioinformatics, some researchers are already entering genomic information in the clinical record. It is also anticipated that, when the cost of sequencing of the full human genome falls, this information will be routinely entered into the clinical record, even though most of that information is currently of no clinical use.Footnote 12 Moreover, there is discussion in the literature as to whether (or how) the findings from genomic research will be fed back to patients. (Clayton & Ross, 2006; Kohane, Mandl, Taylor, Holm, Nigrin & Kunkel, 2007; Miller, Giacomini, Ahern, Robert & de, 2008; Wolf, Lawrenz, Nelson, Kahn, Cho, Clayton et al. 2008) This further blurs the distinction between research and clinical care.Footnote 13 In future, there may be a high level of conflation of research and clinical care, particularly if the direct feedback of research results to patients becomes common practice. (Miller et al., 2008)
2.3 Changing nature of observational research
Much of the observational research described above is shifting from data collection for specific time-limited, clearly defined studies to the development of databases (registries), biobanks, and combinations thereof that will serve as platforms for a wide variety of research undertakings. Registries were initially developed either to facilitate the identification and study of patients with relatively rare health conditions or with treatments that ran a high risk of potential adverse health effects. In recent years, they have begun to proliferate in response to the challenges associated with conducting multi-site observational studies. Two decades ago, these registries held little more than diagnosis and contact information. Now they tend to be very data-rich and minimize the need to go back to the clinical record. The size of the registries varies considerably. Some registries are consent-based, some work by means of notification and opt-out, and some offer no opt-out.
Currently, there is no systematic documentation of the various research registries and biobanks in existence in academic research centres. Moreover, it is unclear what the lines of accountability are for these holdings. Research ethics boards are generally responsible for reviewing research proposals. It is not clear whether ongoing monitoring of the function of registries falls within the purview of REBs. (Gibson, Willison, Brazil, Coughlin, Emerson, Fournier et al. 2008)
Under the Fair Information Principles, a researcher should collect only as much information as is required to accomplish the intended research question. When the goal is to develop a research platform to answer future unforeseen research questions, it is not possible to satisfy this requirement. A long-range solution is required to address this.
The common interoperable EHR is unlikely to eliminate the need for research registries. Data extracted from health records generally require cleaning and re-organization before they are research-ready. (Tezeta, 2008) Also, registries may be augmented with data collected specifically for research purposes.
2.4 Consent-related challenges
Consent raises a number of challenges in observational health research using existing data. Below, these challenges are framed in the contexts of use of existing data with no contact where the identity of the individual is immaterial, and where the researcher requires the information to contact individuals.
2.4.1 Exemptions from requiring consent
While exemptions for seeking individual consent may be obtained for release of personal health information to a researcher, the Tri-Council Policy Statement requires that the researcher demonstrate to the REB that identifying information is essential to the research, adequate safeguards are in place to protect the identity of the individual, and individuals whose data are to be used have not objected to that use. In addition, the laws in many provinces require the researcher to demonstrate that it is impracticable to obtain consent and some call for a weighing of public interest in the research. Finally, in most cases, it is legally required that disclosure of this information is on condition that the researcher will not attempt to directly contact those individuals. There is considerable scope for interpreting when it may be impracticable to obtain consent. To assist in this interpretation, the CIHR’s Privacy Best Practices document states that obtaining consent may be impracticable where there is difficulty in contacting individuals due to:
- the size of the population researched,
- the proportion of individuals likely to have relocated or died since the data were originally collected, or
- the lack of existing or continuing relation between the individual and the organization.
If, because of the above, there is a risk of introducing substantial bias into the research, or if the additional financial, material, human, organizational and other resources impose such a burden as to not permit the research to proceed, then this is considered reasonable grounds for demonstrating impracticability. (Canadian Institutes of Health Research, 2005)
There is evidence that, even with this clarification, there is still considerable disagreement across REBs as to whether consent is required for research involving retrospective review of medical records. (Willison, Emerson, Szala-Meneok, Gibson, Schwartz, Weisbaum et al. 2008a). This reflects a broader challenge of inconsistency in the ethics review process and judgements across research ethics boards more generally. (Subgroup on Procedural Issues for the TCPS (ProGroup), 2008)
2.4.2 Impact of consent on validity of health research
As suggested above, there is growing evidence that selection bias may result from the requirement to obtain consent to use one’s personal information for health research. (Kho, Duffett, Willison, Cook & Browers, 2008) The nature and direction of the biases observed are inconsistent, making it difficult to predict the impact. Of particular concern is differential participation in ways that affect conclusions about health outcomes. For example, with the Canadian Stroke Registry, people with the most severe strokes and with less severe strokes or transient ischemic attacks were less likely to participate in the study. In the former case, recruiters were either reluctant to approach families in their acute illness, or the patient died before they could be approached. In the latter case, many patients were discharged from the hospital before recruiters were able to contact them and several hospitals did not allow the recruiters to contact the patients once they left the hospital. (Willison, Kapral, Peladeau, Richards, Fang & Silver, 2006)
2.4.3 Limitations of existing consent models in the context of registries and biobanks
Where it is determined that consent is required for participation in a research registry, there are challenges with satisfying existing consent requirements. These have been framed around the needs for clinical trials where:
- There is a single study with specific research goals;
- One can specify in advance data elements required for analysis;
- Risks of harm associated with participation are readily quantifiable.
As described in Section 2.3, the nature of observational research is changing from traditional studies bounded by specific research questions to the creation of research platforms. This is pressing the limits of the traditional consent process. In particular, it is not possible to describe every possible research use of the information, as many of the future research questions that may be posed are unknown. This raises the question of how much detail is sufficient to satisfy the requirement for a fully informed consent. If too little information is offered (e.g. a blanket consent for “all future research uses”) then one could justifiably argue that the consent was not valid. If a description of every conceivable future use was required, then this could well obfuscate the key information that would inform the individual as to what she was committing to and would likely still fall short of providing complete information. For a more complete discussion of these issues, the reader is directed to the recent work of Kosseim and Brady. (Kosseim & Brady, 2008)
Given the many potential research uses of one’s health information, recent discussion has moved away from the dichotomous choice of project-specific consent vs. exemption from any consent requirement to that of tiered consent. (Caulfield, Upshur & Daar, 2003; Singleton & Wadsworth, 2006) Research suggests people’s consent choices are influenced by the type of research, who is conducting it, the type of information to be accessed or linked, and the degree of commercial element to the research. (Willison, Schwartz, Abelson, Charles, Swinton, Northrup et al. 2007; Willison, Swinton, Schwartz, Abelson, Charles, Northrup et al. 2008b) Generally, most people find a passive consent process (notification with opt out or use without notification) acceptable for “public good” uses like quality improvement and public health, but desire greater control when the research involves the potential for commercialization of products of discovery. However, most research cannot be cleanly categorized into pure public good research or pure commercial.
If offering multiple consent options for a variety of research uses of one’s health information, one must take care to avoid decision fatigue. (Ram, 2008) This challenge is particularly germane in the context of registries and biobanks, and will become even greater with the advent of whole genome research and translational bioinformatics.
2.4.4 Contacting individuals to actively participate in research
A major challenge in health research is the identification and contacting of individuals to seek their participation in health research, whether for an observational study or clinical trial. In the United States and in the United Kingdom, one explicit objective of implementation of the EHR is to assist in the recruitment of patients for clinical research. (Anonymous, 2007a; Anonymous, 2008a; Westfall, Mold & Fagnan, 2007) While this has not yet surfaced as a future use of the EHR in policy discussions in Canada, this should be anticipated.
Current practice in Canada requires a two-stage consent process, wherein:
- Patients are initially approached by someone who has reasonable expectation of access to information about their health condition and who is not in a position of undue influence. The purpose of the initial approach is to apprise the potential research participant of the research project and to either obtain their permission to release their name and contact information to the researchers, or to provide the individual with the contact information of the researcher whom they may call if interested in participating in the research.
- Those interested in being contacted discuss the specifics of the study with a member of the research team and the patient makes a free and informed decision whether or not to participate.
While the CIHR Privacy Best Practices document articulates current best practice in this area, this process often results in substantial loss of potential research participants, and ensuing selection biases. Previous research has found the public to be more comfortable with a trained research assistant from an academic centre screening their records for study eligibility than they were the doctor’s secretary. (Willison et al., 2007) Also, in light of lobbying by researchers in British Columbia, the new e-Health (personal health information access and protection of privacy) Act has created provision for directly contacting the patient, subject to the approval of the provincial Information and Privacy Commissioner. (Abbott, 2008)
2.4.5 Lock-box provisions in some legislation
In some provinces, there are “lock-box” provisions in legislation that allow patients to control who may see particular information in their record. They are generally intended for application in the context of direct healthcare provision, where individuals could limit who may have access to information in the record that they found particularly sensitive.
It is not clear whether the lock-box provision would apply for research where the identity of the individual is immaterial, the data are de-identified prior to research use, and where there will be no attempt to contact that individual. Under these conditions, use of that information for research would appear to be at minimal risk.
As mentioned above, independent of lock-box provisions, researchers are generally not permitted to directly approach patients based on some prior knowledge of their health condition, if there is no prior relationship with that individual. Screening and initial contact are generally carried out by someone with reasonable expectation of access to this information. In this case, it would make sense for lock-box provisions to apply to the screener whose role is to identify potential research participants, based on data available in the clinical record.
2.5 Research culture
2.5.1 International initiatives to improve access to research results
Historically, the culture among researchers – particularly those who work in epidemiology, public health, health services and policy research, population health, and quality improvement – has been to make publicly available the findings from their research at the earliest possible time. It has also been common to make raw data available to other researchers to verify earlier published results, to test a new research question, or to compile information from across several studies to look for overall patterns of outcomes, where the smaller studies may have had insufficient statistical power to do so. This is now general policy of major public granting agencies internationally, including the National Institutes of Health (NIH) in the United States, the Medical Research Council (MRC) in the United Kingdom, and the Canadian Institutes of Health Research (CIHR). (Canadian Institutes of Health Research, 2008; Medical Research Council, 2008; National Institutes of Health, 2008)
Increasingly, this open access has been accomplished through posting of de-identified data on a website. This is a particularly common practice in genomics research. Since the ability to re-identify data that were previously considered sufficiently anonymized is constantly evolving, the practice of openly posting of raw research results online is questionable. A recent case in point was the re-identification of individuals in pooled genetic data that were posted online. (Couzin, 2008)
The culture among researchers of open data sharing fits poorly with the Fair Information Principle of limiting disclosure of personal information. It also exists in tension with researcher motives to retain control over the data. There is a competitive advantage associated with restricting access to a dataset that may yield new research for the possessor of that information. This is particularly the case if there is potential for intellectual property protection and commercialization. (See Section 2.5.3 below.)
2.5.2 Data retention
In general, researchers are reticent to destroy data. They are acutely aware of the cost of data collection – particularly the person-hours of staff and volunteer patients – and desire its re-use for other purposes, wherever possible. In addition, data need to be retained for a variety of purposes, including:
- The possible need for re-analysis, should findings be disputed. One cannot simply “cut” another dataset from the original source, as there is considerable work in preparing raw data for research use and, inevitably, raw datasets are revised over time (e.g. revisions to data at the source) so that it becomes almost impossible to completely reproduce a dataset;
- The need for audit trails, in the event of allegations of fraud, including fabrication of data.
This reticence to destroy data is in tension with the Fair Information Principle of limiting the duration of retention of personal information. Instead of data destruction, researchers prefer solutions involving long-term secure storage.
2.5.3 Commercialization of innovations and intellectual property
The introduction of the Bayh-Dole Act in the United States in 1982 marked a tide-shift internationally in the commercialization of innovations discovered using public funds. (Kennedy, 2005) Prior to this, discoveries using public funds were considered to be in the public domain. There was concern that insufficient attention was being given to the uptake of these innovations. The proposed solution was to actively promote the commercialization of these innovations with intellectual property rights being awarded to the institution and the researcher responsible for the innovation. There has been considerable controversy over whether the net effect on innovation has been positive or negative. (Boettiger & Bennett, 2006; Eisenberg & Nelson, 2002; Thursby & Thursby, 2003)
This issue is germane to privacy and research use of information from the electronic health record for several reasons. The introduction of a commercial element to research causes people to desire greater control over use of their health information for research. (Willison et al., 2007; Willison et al., 2008b) One of the most intensive research applications of the interoperable EHR will be in the area of translational bioinformatics, where there will be a high level of commercialization of innovation. Historically, there has been relatively little interest in commercialization of discoveries of research in public health, health services and policy, quality improvement, and population health. However, in recent years, the application of intellectual property rights has extended to particular research methods. (Classen, 2006; Walker, 2006) This may become increasingly the case as genomic researchers delve into the relationship between genetic and environmental facts in affecting health. (Anonymous, 2008d)
2.6 Governance issues
Governance over research use of health information can be thought of as occurring at three levels: (1) review of individual projects (micro-level), (2) management of research repositories (meso level); and (3) overall governance of research uses of health information generally (macro level).
2.6.1 Review of individual research projects
A study of the responses of REB chairs and administrators to scenarios involving medical record review and the development of registries and biobanks revealed a high variation in opinion regarding the need for consent for research involving medical record review, and some uncertainty on the part of REBs over whether their purview extended to oversight of the establishment and function of registries. (Gibson et al., 2008; Willison et al., 2008a) It should be noted, though, that this study was carried out at a time when several of the provinces were introducing or revising their data protection legislation and the CIHR had just developed its Best Practices for the Protection of Privacy in Health Research. (Canadian Institutes of Health Research Privacy Advisory Committee, 2005) At this time, there was still high uncertainty as to what was and was not permitted. Anecdotal reports suggest this issue persists.
2.6.2 Management of research repositories
Academic health research is conducted in a wide variety of settings. The greatest volume of research is conducted in a handful of large data institutes affiliated with major universities. These institutes are relatively well-resourced with the capacity to implement and manage procedural and statistical approaches to data disclosure control. However, a large number of smaller research teams exist in universities and academic healthcare facilities. They vary greatly in size from one researcher with a few research assistants and students to clusters of researchers who may share research staff. The capacity to securely manage data access and disclosure varies greatly, including policies on how to archive data once the research is complete.Footnote 14
The larger data institutes currently undergo periodic auditing of their data management practices. The weak point in the system lies with the large number of smaller holdings based in universities and academic healthcare facilities. Currently, there is no routine audit of data management practices in these settings. Within most of these facilities, there is no centralized support system for the secure management of research data. While the institutions generally have policies on data management, each research team within the institution is responsible for the development and maintenance of its datasets and for security. Attention to these details is uneven. An example of the consequences of such a decentralized system of accountability is chronicled in the report of the Ontario Information and Privacy Commissioner regarding the theft of a laptop computer with identifiable data from multiple research projects. (Cavoukian, 2007)
As the interoperable EHR comes into effect, there may be increased opportunity for the establishment of smaller research data repositories that are poorly regulated. Under the current disclosure-based system, transfers of data can be reasonably tightly controlled. The forthcoming interoperable EHR will be an access-based system, under which it may be possible for those with permission to access records for clinical purposes to screen records with a view to developing their own ad hoc research databases within their institutions and even pool those data with colleagues at other institutions, unless specific access controls are introduced.
2.6.3 Overlapping jurisdiction in the governance of research uses of health information
Oversight of the development and use of research repositories falls within the purview of several bodies. On the one hand, oversight of the protection of privacy of Canadians falls clearly within the realm of privacy commissioners/ombudsmen. Whether this would fall under federal or provincial legislative authority is unclear. Most of the provincial data protection laws and the federal Personal Information Protection and Electronic Documents Act (PIPEDA) have clauses regarding research uses of personal (health) information, but the direction is very general and, for the most part, leaves the academic community to work out its own processes. PIPEDA exempts some academic research from requiring consent for use of data collected through third parties. One of the conditions is that the organization informs the federal Privacy Commissioner of the use before this information is used, but this condition has never been exercised. Section 39 of the Ontario Personal Health Information Protection Act provides for a handful of prescribed registries subject to the oversight of the Information and Privacy Commissioner of Ontario. An expansion of oversight to all registries in the Province would overwhelm the Office.
Privacy protection of research participants is also a component of human research protection. In Canada, there is no specific legislation. Instead, human research protections are informed by the Tri-Council Policy Statement on Ethical Conduct for Research Involving Humans. (Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada & Social Sciences and Humanities Research Council of Canada, 2005) Nor is there currently an accreditation body for protection of human research participants. In Canada, discussions have been underway for some time to implement a program of accreditation of institutions’ human research protection programs. (The Experts Committee for Human Research Participant Protection in Canada, 2008) One could argue that an important part of that accreditation program should be auditing the consistency of institutions’ information management practices with current standards.
From yet another perspective, one could argue that research uses of information from the EHR should fit within a larger health information governance structure, similar to that of the National Information Governance Board for Health and Social Care under the National Health Service in the UK. (Anonymous, 2008b)
The risk with multiple overlapping jurisdiction in this matter is not that there will be competition over who will take the lead, but that there will be a vacuum in leadership as each body looks to the other to take the lead.
2.7 Possible loss of source data
As described in Section 1.2, some health research requires access to information that resides outside the health record. In cases where linkages need to be made between health information and information external to the health record, there is the risk that the external information may have been destroyed in compliance with data protection laws requiring that data be retained only as long as required for their intended purpose. This is best illustrated by an example from occupational health and safety.
To determine whether a particular cancer may be due to exposure to a particular workplace chemical, it may be necessary to ascertain levels of workplace exposure through employee records. However, there may be a latency period of 20 or more years between exposure and outcome. Therefore, it may be decades after an employee has left the service of the particular firm before any suspicion is raised between exposure to the chemical in question and the development of cancer. If the duration of retention of employee records beyond the period of employment is low – e.g. 5 years – then these data may be unavailable in future for such research.
When data retention policies are being established in workplaces that use potentially hazardous materials, care needs to be taken to ensure data are retained for a sufficiently long period to account for such future potential uses. The chief challenge is that the hazards are not just chemical (e.g. workplace stress) and one cannot anticipate what environmental hazard, broadly speaking, will result in some future illness 10 to 20 years down the road. This is an issue that cannot be easily addressed in the presence of either laws or policies limiting the period of retention of data to that needed for the initially stated purpose.
2.8 Lack of consensus among affected parties
The issue of research uses of health information has been debated amongst researchers, policy makers and privacy advocates for many years. Despite this, many of the parties’ positions have changed little. In 1999, Freeman and Robbins chronicled the (then) 25-year history of debate over privacy and automated collection and storage of personal health data in the United States. (Freeman & Robbins, 1999) They noted repeated failures to come to a working consensus on how to manage personal health information in a way that would protect privacy while allowing for a variety of administrative uses of that information. One concern they identified was poor communication among the stakeholders and a failure of stakeholders to pinpoint where they agreed or disagreed about how the data ought to be managed and controlled. While Canada’s history in this regard has been less confrontational, consensus is still lacking on several issues of control over and access to health information for research and planning purposes.
Substantial inroads have been made already at addressing the conditions under which personal information may be used for health research. In particular, the CIHR Ethics Office has laid considerable groundwork that culminated in their internationally cited Best Practices for Protecting Privacy in Health Research. (Canadian Institutes of Health Research Privacy Advisory Committee, 2005) In addition, the working group on Harmonizing Research and Privacy has pulled together suggested standards for managing confidentiality and security of data. (Slaughter, Collins, Roos, Weisbaum, Hirtle, Williams et al. 2006) Nonetheless, there is still considerable need to build on these developments and identify areas of convergence among the multiple affected parties as to the circumstances under which personal information may be used for health research.An ongoing challenge is that the number of parties with a voice in this issue is large – researchers, ethicists and lawyers, physicians and other data custodians, governments, patients, and family members. This slows progress in effecting change, particularly if interactions promote entrenchment in positions.
2.9 Summary
To summarize, several challenges in addressing privacy in the use of personal information for health research have been identified. Most research relies on person-level data of sufficient detail as to require that the data be treated as potentially identifiable. It is becoming increasingly difficult to distinguish research from other secondary uses of health information that do not require individual patient consent and, in some cases, even difficult to distinguish from clinical care. With shifts in how research is being conducted – moving from discrete studies to registries and biobanks that will serve as research platforms, and the wide range of possible uses – the conventional approach to consent is inadequate. The proliferation of health information holdings along with heterogeneous data management policies raises concerns over the long-term secure management of health information. It also highlights the need for oversight over these collections. As well, there will be increasing pressures from researchers for more direct access to patients for purposes of contacting them to participate in research. The current two-stage mechanism of initial contact with patients does not work well, yet it’s alternative – direct contacting of patients by researchers with no reasonable expectation of access to their personal health information – raises major privacy concerns and may contravene current privacy laws. Together, these issues call for more than change at the margins or for an attempt to bar access to data. Instead, they call for a fundamental re-consideration of how information is used for research and the role of research vis-à-vis clinical care, public health, and quality improvement. We need to consider what a coherent information management system would look like if we were to begin de novo, knowing what we now know.
3. Potential Approaches to Addressing Research Uses of Personal Health Information in the Context of the EHR
3.1 Re-thinking the role of research vis-à-vis other secondary uses of PHI
Academic research occupies a different status from quality improvement, public health, and program planning, which are generally exempted from requiring patient consent and from ethical scrutiny. While historically this may have been justified, the practice of quality improvement, systems planning, and public health use common epidemiologic tools and expose patients to the same level of risk as research using the same techniques. So, these boundaries are now artificial and should be dissolved. This appears to be the approach taken in the Care Record Guarantee in England, which describes the uses of the care record as follows: (National Information Governance Board, 2007)
The people who care for you use your records to:
- provide a good basis for all health decisions made by you and healthcare professionals;
- allow you to work with those providing care;
- make sure your care is safe and effective; and
- work effectively with others providing you with care.
Others may also need to use records about you to:
- check the quality of care (such as a clinical audit);
- protect the health of the general public;
- keep track of NHS spending;
- manage the health service;
- help investigate any concerns or complaints you or your family have about your healthcare;
- teach healthcare professionals; and
- help with research.
It has been argued that a proportionate approach to ethical review should be applied to both routine quality improvement activities and quality improvement research, according to the degree of risk to individuals. (Alberta Research Ethics Community Consensus Initiative, 2005) This would dissolve the artificial distinction between research and routine monitoring activities in these areas, and even raise the ethical bar for quality improvement activities. Similar arguments may be made in the areas of public health and systems planning. It would be necessary to develop and adequately resource the ethical review mechanisms and ensure reviewers are adequately trained, so as not to bog down these quality improvement, systems planning, and public health activities.
Frameworks for guiding the proportionate review process need to be developed. In quality improvement, the Alberta Consensus Initiative developed some frameworks. However, these have been criticized for being overly complex. Another framework for privacy-related risk-analysis associated with research is that of Chamberlayne and colleagues. (Chamberlayne, Green, Barer, Hertzman, Lawrence & Sheps, 1998) (See adaptation in Figure 1.) This framework was developed in the context of large database analyses. In addition, the CIHR Best Practices for Protecting Privacy in Health Research provides a useful conceptual framework. (Canadian Institutes of Health Research Privacy Advisory Committee, 2005)
Given the longstanding secondary use of health information for quality improvement, systems planning, and public health, consideration should be given to expanding the primary uses of health information to encompass the management of (a) the health of individuals; (b) the health of populations; and (c) the health care system.
This conceptual shift would then acknowledge quality improvement, public health, population health, and systems planning activities as legitimate primary uses of the data, and the common interoperable EHR would represent a rational approach to managing this expanded purpose. By extension, research that supports one or more of these three primary uses of the EHR would be considered a primary use.Footnote 15 This is consistent with the original framework for the EHR put forward by the Advisory Council on Health Infostructure. (Advisory Council on Health Infostructure, 1999) In addition, the blueprint for the common interoperable EHR has already been revised to accommodate public health uses of the EHR. (Canada Health Infoway, 2006)
Designation of certain types of research or evaluation activities as primary uses of the EHR would not obviate the need for ethical review, nor does it dispense with the question of whether individuals could opt-out of some research uses of their health information. (See Section 3.2 below.) What it would do is provide a conceptual shift that acknowledges the legitimacy of research uses of personal information under prescribed conditions. This is a concept that would require an open public deliberation process and, if adopted, public education over time. It may also obviate the need for project-specific consent.
RECOMMENDATIONS:
- The artificial boundaries between research in and the practice of quality improvement, systems planning, and public health should be dissolved.
- Research uses should be treated equally to these other secondary uses and ethical review should be applied to all these secondary uses, proportionate to the risks associated with these data uses.
- Practical frameworks need to be developed for evaluating risks and benefits of these secondary uses of health information.
- In the long run, consider re-conceptualizing the primary uses of personal health information to include the management of the health of: individuals, populations, and the health care system. Research supporting these activities would also constitute primary uses. Such a shift would call for open public deliberation.
Figure 1. An approach to evaluating privacy risk in database research
(Adapted from Chamberlyne et al, 1998)
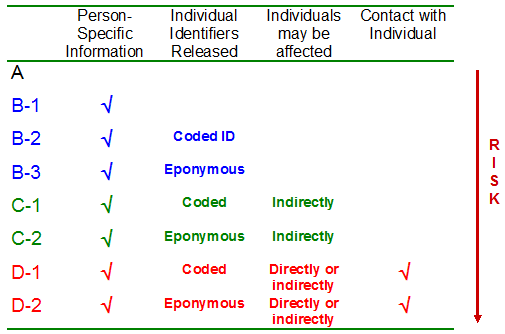
3.2 Consent
3.2.1 Re-thinking the role of consent
In the context of research involving genetic information, O’Neill has argued that several shortcomings to the consent process make our conventional view of valid consent an impossible cognitive feat, even for individuals who have full decisional capacity. (O'Neill, 2001) She argues that the information is complex and opaque with so much detail that no one has the ability to either explain or comprehend it completely – even the researchers. In addition, at the time of presenting in the health care system, individuals are highly stressed and vulnerable. She argues that, in the process of shifting emphasis in ethics from paternalism to autonomy, the writing in medical ethics has placed too much weight on human capacities to take in and understand complex information. Moreover, she points out that the context of consent has changed substantially, with research being conducted in an environment of a complex array of information and regulatory systems. Yet the consent model still operates largely on the assumption of a one-to-one relationship between the researcher/clinician and the patient in an environment where mutually respecting physician and patient can openly converse, weigh risks and determine the best course of action. The same arguments could be used for the many other potential research uses of information from the electronic health record.
In light of these shortcomings, O’Neill has argued that we need to move from individual consent for particular acts of collecting, using, or storing data to a “public consent” (i.e. mandate) to systems for collecting, storing, using, and disclosing of data. (O'Neill, 2001) She points outthatthis reduces the demands on individual consent procedures, but does not obviate entirely the need for individual consent. Unfortunately, in the context of this public mandate for systems of information use for research, O’Neill does not articulate what that individual consent should involve. In the next few paragraphs, suggestions are made as to what such a system might look like and how the public – individually and collectively – could be involved in governing use of their personal information for health research.
3.2.2 Consent choices
Much of the literature addressing consent for research use of one’s information has taken a dichotomous approach to consent, where either conventional project-specific consent applies or the research is exempted from requiring consent under specified conditions. Project-specific consent is impracticable for much of the observational research being considered in this report. On the other hand, the ability to say “No” to certain types of research greatly increases the confidence of the public in the use of their information for health research. (Willison et al., 2008b)
Recent discussion in the literature has taken a more expansive approach, acknowledging that consent could be tiered and may differ depending on the particular type of research and other factors. (Singleton & Wadsworth, 2006)For example, one could be offered a menu of choices:
- Not using one’s information for a particular type of research;Footnote 16
- Requiring permission each time before that information is used;
- Requiring general opt-in permission for a particular type of research, with the ability to apply restrictions;
- Requiring notification with the option to opt-out;
- Permitting use without notification.
There are many possible choices that individuals could be asked to make regarding secondary use of their personal information – whether for research or for other uses. If given the full spectrum of choice for each use, one runs the risk of “decision overload”, which could cause individuals to make arbitrary choices, refrain from choosing altogether, and experience regret following decision-making. (Iyengar & Lepper, 2000; Ram, 2008) Therefore, when designing a system for ascertaining consent choices for use of people’s health information, one needs to be mindful of this risk.
3.2.3 Suggested ways of eliciting, documenting and communicating consent choices
Eliciting and documenting the consent choices of individuals is not a simple process. Any system will take time to design and implement, and will require an adequate time for both informing about choices and making those choices. It is unrealistic to expect that this could be accomplished through one’s family physician. Physicians simply haven’t the time to take on this role and a growing proportion of the population have no regular family physician. One should expect to spend at least 30 minutes for information to be conveyed and choices to be made – even longer, if offering a menu of consent options across a variety of uses of one’s personal health information. In Quebec an attempt to broker a simple consent for use of an electronic health record through the family physician failed because the average 1.5 minutes taken to obtain the consent was considered by most physicians to be excessive.Footnote 17
Informing and brokering:
A variety of approaches and settings should be used to match the needs of individuals with appropriate technologies. For example, one could prepare explanations of the uses of one’s health information both in brochures and on DVDs to be played in hospital clinics, physicians’ waiting rooms, or the public waiting area of Ministry of Health offices where people come to renew their health card. Manson and O’Neill point out the importance of the process of informing and not just transferring information to the individual – i.e. the information brokering role. (Manson & O'Neill, 2007) It is an important step in building trust and understanding. Therefore, a knowledgeable individual – e.g. a nurse in the clinic – should be available to answer any questions or concerns an individual may have, and space made available for completing the consent process. Much of this also could be done through secure websites at one’s own leisure. Again, there need to be opportunities to talk directly with individuals who are knowledgeable of the system and the options available.
Documenting and communicating choices:
In the context of the common interoperable EHR, much of the eliciting, documenting and revising of one’s consent choices could be accomplished through an electronic patient portal to one’s own health record, as part of one’s personal health record system. (Grant, Wald, Poon, Schnipper, Gandhi, Volk et al. 2006; Hess, Bryce, Paone, Fischer, McTigue, Olshansky et al. 2007) This would enable individuals to manage and communicate, in an ongoing fashion, their consent choices for research and other uses of their personal health information (including the equivalent of “lock-box” by indicating that information cannot be used for particular purposes.) It would also provide a way of being able to review what types of research have been conducted using their health information. This could be accomplished by linking with data from the registry of research data holdings described in Section 3.3. It would also provide a place for posting a notice of information use practices equivalent to the NHS Care Record Guarantee. (National Information Governance Board, 2007) The patient portal would give people the opportunity to indicate whether they wished to have research results fed back to them and a way of receiving those results. (Kohane et al., 2007)
One way of simplifying the choice process to minimize decision overload is to assign different default consent options for different types of research, with provisions (in most cases) for individuals to modify the default option to either a more permissive or more restrictive consent option. These default positions would need to be determined through a deliberative process involving researchers, ethicists, regulators, and the public. Drawing from earlier public engagement work (Willison et al., 2008b), a hypothetical sample grid of what this might look like may be found in Table 3. Recognizing the safety benefits to individuals and the public, the default position for public health and the monitoring of new drugs and devices for safety may be mandatory use of this information with no option to opt-out. For quality improvement and population health research, the default may be notification with the option for individuals to opt-out. The rationale here is that this is minimal risk research with no patient contact. The opt-out provision provides at least some degree of control in the hands of those whose data are being used. Historically, few people opt-out (in the range of 1 in 1000 to 1 in 500) and the impact of such a low opt-out on scientific integrity is minimal.
For research involving biological samples linked with personal information from the health record, the default may be a broad opting in with the option for individuals to opt for project-specific consent or to specify particular restrictions on the types of research that could be conducted. The switch to an opt-in default for this kind of research reflects the greater intrusiveness (and potentially invasiveness) of research involving biological samples.
In a similar fashion, one could use the patient portal for people to indicate their willingness to be contacted to actively participate in specific types of research activities – for observational studies or clinical trials. Those interested could then specify whether it would be acceptable for the researcher to contact them directly or if they wished to be contacted through their family physician or other care provider.
There are numerous logistical challenges to consider, including:
- Equity in access to mechanisms for expressing one’s consent choices (e.g. through patient portals to their EHR);
- How to manage expectations of just how much control will be offered through the patient portal;
- How to ensure that the opting-out process is not done capriciously;
- The impact of commercialization of products of innovation on consent choices.
All of these require further research.
RECOMMENDATIONS:
- It has been suggested that we move from individual consent for particular acts of collecting, using, or storing data to a public mandate for systems within which data are collected, stored, used, and disclosed. (O'Neill, 2001) Such a move will require public deliberation and consensus development.
- This public mandate does not obviate entirely individual choice. At the individual level, we should consider a more expansive approach to consent that includes broad opt-in to a program of research and notification with opt-out.
- Mechanisms need to be developed for both eliciting and documenting in advance individual consent choices for different types of research uses of one’s personal information. Suggestions for this are offered in the text. The same process could be used to determine people’s preferences for how to be approached for participation in research studies.
- To minimize decision overload, default consent choices are suggested for different types of research uses. For example, for public health and post-marketing surveillance of drugs and devices (whether or not formally defined as “research”), the default may be mandatory inclusion of one’s data. For quality improvement and population health research, the default may be notification with opt-out. For studies involving biological samples, the default may be a broad opt-in. (See Table 1 for details.) Again, the default consent choices should be determined through a public deliberative process.
- Research is required into logistical challenges, including: equity in access to mechanisms for expressing one’s consent choices (e.g. through patient portals to their EHR); how to manage expectations of just how much control will be offered through the patient portal; how to ensure that the opting-out process is not done capriciously; and the impact of commercialization of products of innovation on consent choices.
| Example of Research Use* | Proposed Default | Proposed Patient Options |
|---|---|---|
| * Note: The examples provided have simplified the role of commercialization of innovation. For example, public health surveillance assumes a pure public good – i.e. no private profit or intellectual property protection. Empirical research suggests that consent choices are very sensitive to the presence of a profit element. ** For transparency, there should also be some sort of public notification of the uses made of this information. *** Where practicable (e.g. once patient portals to their EHRs are established), the notification could be individualized. |
||
|
Public health surveillance, with no individual contact, and no commercialization of innovation |
Mandatory participation** |
None |
|
Post-marketing surveillance of selected new drugs and for devices, with no individual contact and no commercialization of innovation |
Mandatory participation** |
None |
|
Quality Improvement / systems planning, with no individual contact and no commercialization of innovation |
Notification with option to opt-out*** |
|
|
Population health, with no individual contact and no commercialization of innovation |
Notification with option to opt-out*** |
|
|
Research involving linkage of health information with biological samples, whether or not commercialization of innovation is involved |
Opt-in – broad consent |
|
|
Participation in a registry documenting people willing to be contacted to participate in prospective research |
Initial contact must be through someone in the circle of care |
Researcher may make initial contact with the individual directly |
3.3 Context within which research would be conducted
3.3.1 Physical infrastructures
Regardless of whether research uses of health information are considered primary or secondary in nature, infrastructures will be required to enable ethically and scientifically valid research to proceed with adequate safeguards. The basic data architecture for research databases differ from those designed for the provision of care. Therefore, in the context of an interoperable EHR, an adequate analytic capacity requires the maintenance of separate research databases. In part, this is because the design of the EHR is oriented toward real-time transaction-oriented uses in clinical care, which does not allow for robust analytic processing of the data for epidemiologic-type analyses. (Sanders & Protti, 2008) The addition of analytic capacity should be designed in from the outset, as retrofitting often results in poorer quality of data.Footnote 18
Historically, analytic capacity has been poorly managed by governments, so several provinces have made arrangements for these services to be provided through research institutes affiliated with universities – e.g. the Population Health Research Unit in Nova Scotia (PHRU), the Institute for Clinical Evaluative Sciences in Ontario (ICES), the Manitoba Centre for Health Policy (MCHP), and the Centre for Health Services and Policy Research in British Columbia (CHSPR). These centres are operated by academic researchers who, in addition to conducting analyses for their provinces, have conducted their own academic research. In effect, they act as data custodians, similar to hospitals. Provisions are made in provincial laws to permit the special status of these institutes. (Kosseim & Brady, 2008) At these centres, raw data are cleaned, linked, stripped of direct identifiers and made research-ready, using strict protocols to limit access to identifiable data to a limited number of individuals. There are also ethics review mechanisms in place and periodic external audits of data management practices.Footnote 19 (Slaughter et al., 2006)
At ICES and MCHP, raw data never leave the institution (enclave model). By contrast, at CHSPR, data are selectively released to meet the needs of the particular research request and provisions are made in the user agreements for raw data to be destroyed at some pre-specified point in time. In the enclave model, access to data for research is restricted by geographic and other membership limitations. This is a highly secure model for data management. In exchange, there are geographic inequities in access to data. In Ontario, this regional inequity in access is being addressed through the development of dedicated satellite sites that have direct “hard-wired” access to ICES servers and rigorous safeguards in place.
Where data are released directly to researchers, access is greatly improved, but controls on raw data use, once released, are potentially weaker, since one is relying upon third parties to maintain safeguards. (Black.C, McGrail, Fooks, Baranek & Maslove, 2005) Secure release of data directly to the researcher could be possible, with use of digital rights management technology embedded in the data released. In that way, limits could be placed on the computers on which the data may be loaded, and on the duration of authorized use. Recognizing that identifiability exists on a continuum, El Emam has suggested that custodians customize the extent of de-identification of data prior to their release, subject to the extent of controls that the recipient has to safeguard these data. (El Emam, 2008) This presupposes that data custodians have readily-available techniques to customize the level of identifiability of released data – for example, through reduction in detail (e.g. replacing date of birth with year of birth) and random perturbation of the data. (El Emam et al., 2007)Footnote 20 Currently, these disclosure control and limitation techniques are not in general use among data custodians. Implementation of these technologies and training of personnel in their use should be encouraged.
RECOMMENDATIONS:
- The practice of directly posting raw data online – even if de-identified – should be discouraged. Instead, the existence of these repositories could be posted online with mechanisms to ensure the data could be available to researchers for bona vide research purpose, with appropriate disclosure controls and user agreements in place.
- Secure data repositories should continue to be developed, with conventional access and disclosure control mechanisms in place. (Jabine, 1993; National Research Council, 1997)
- New technologies that enable secure remote access to these repositories should be adopted and both data custodians and researchers should be trained in their use.
3.3.2 Governance
Review of individual research projects:
Section 2.4.1 described the challenge of variation across REBs in judgments regarding the conditions necessary to permit research use of personal health information without consent. One approach to addressing this is to develop a set of case-based tutorials for REB members, data custodians, and for researchers. This could build on and refine the case studies developed by the CIHR in 2002. (CIHR working group on case studies, 2002) In the case of research involving the common interoperable EHR and biological samples, the privacy and confidentiality-related issues may be sufficiently technical as to require expertise from centralized specialty review bodies that have the capacity to review research protocols involving access to data from the EHR and use of biological samples.
Managing research repositories
The proliferation of research repositories could be managed by requiring (e.g., through legislation or regulations) the approval and registration of all research registries, databases or biobanks with the province, subject to:
- Justification of why existing data repositories could not be used;
- Meeting standards for managing privacy, confidentiality and security, and governance;
- Adequate training of staff and researchers;
- Periodic audit of information use practices.
Wherever possible, data and sample repositories should be under the stewardship of the institutions rather than individual researchers. The institution would then be accountable for secure storage and management of the repositories and for enforcing access controls and other safeguards. This would also ensure that REBs had been consulted, as appropriate, before data were accessed or released for specific research projects. It would also permit the ongoing collection and maintenance of rich datasets by the institution while satisfying fair information principles on limiting data “collection” by instead limiting the amount of data either accessed by or disclosed to the researcher, consistent with that required to accomplish the intended research. With the registration of databases and biobanks, it would then also be possible to keep track of particular studies that are being conducted.
Governance over research uses of health information generally
Given the current state of flux around accreditation of human research protection programs in Canada and the absence of research uses in Infoway’s blueprint, there is a leadership vacuum in governance at this level. The federal and provincial privacy commissioners/ombudsmen should consider whether they wish to take a lead in this regard. If so, actions should be carried out in consultation with the other potential governors of these research uses of health information.
Regardless of who becomes the chief oversight body for privacy protections, one important element is to require institutions to maintain an inventory of:
- the databases and biobanks within their organization;
- the research studies that emanated from the databases; and
- any disclosures of the data to other institutions (including a centralized repository).
Reports could be filed annually to the prime regulatory body responsible for privacy and data protection matters, with access to this information by the other regulatory bodies. These research institutions would then be subject to periodic audit by the prime regulatory body, and reports of these audits circulated to the other governing bodies and made available to the public. This regulatory body should also be available to data custodians in a consultative and advice-giving capacity, in a spirit of continuous quality improvement.
Managing “information usage creep”
Finally, the research repositories developed can be very potent analytic tools. Thus, the very features of these repositories that spark great anticipation on the part of researchers also increase the potential for invasion of privacy. When developing the research infrastructure, one needs to consider how best to scrutinize and to limit uses that are inconsistent with the original research purposes of the research repository. For example, with regard to the clinical record, there have been concerns over compelled disclosure of health information for employment or insurance applications. (Rothstein & Talbott, 2006) An equivalent concern could arise in the case of compelled releases of data from research datasets for government security surveillance activities like the French Edvige database. (Ozimek, 2008) The examples provided are clear cases of uses beyond those intended uses. Many cases will not be so clear. In Canada, there is currently no equivalent to the certificates of confidentiality that can be issued in the United States, protecting researchers from compelled release of data about research subjects. (Coffey & Ross, 2004; Wolf, Zandecki & Lo, 2004) An open question, though, is whether a certificate of confidentiality would hold up to the USA PATRIOT Act or similar national defence legislation.
RECOMMENDATIONS:
Review of individual research projects
- To improve the consistency in the decisions of REBs regarding the conditions for exemption from requiring consent for research, detailed case studies should be developed and deployed as part of a continuing education outreach to research ethics boards and researchers. These case studies could build on the earlier case study work by the CIHR in 2002.
- In the case of research involving the common interoperable EHR and biological samples, consider developing centralized specialty review bodies that have the capacity to review research protocols involving access to data from the EHR and use of biological samples.
Managing research repositories
- Data and tissue holdings intended for research purposes should be registered with the research ethics boards of the institutions where they are housed. Any research projects that emanate from these holdings should also be collated in this database. With the registration of databases and biobanks, it would then also be possible to keep track of particular studies that have been conducted using these data sources.
- Wherever possible, data and sample repositories should be under the stewardship of institutions rather than individual researchers. In this way, long-term secure management of research data may more readily be assured.
Governance over research uses of health information generally
- Given the current leadership vacuum in governance at this level, the federal and provincial privacy commissioners/ombudsmen should consider whether they wish to take a lead in this regard. If so, actions should be done in consultation with the other potential governors of these research uses of health information.
3.4 Engaging all affected parties, including the public
Substantial inroads have been made already at addressing the conditions under which personal information may be used for health research. In particular, the CIHR Ethics Office has laid considerable groundwork that culminated in their internationally cited Best Practices for Protecting Privacy in Health Research. (Canadian Institutes of Health Research Privacy Advisory Committee, 2005) In addition, the working group on Harmonizing Research and Privacy has pulled together suggested standards for managing confidentiality and security of data. (Slaughter et al., 2006) There is need to build on these and other accomplishments to identify areas of convergence and divergence among all affected parties on the conditions under which personal information may be used for health research. This would best be addressed through a series of evidence-informed deliberative dialogues among the full spectrum groups involved with or affected by the use of personal information for health research.
The general public have been outside these discussions. They generally have a low awareness of just what kinds of research are currently being conducted using personal health information and what future uses are being contemplated. (Robling, Hood, Houston, Pill, Fay & Evans, 2004) Some public engagement has been conducted to date in Canada and internationally regarding policy making in these areas. (Damschroder, Pritts, Neblo, Kalarickal, Creswell & Hayward, 2007; Robling et al., 2004; Willison et al., 2008b)
As we move toward more operational models of use of personal health information for research (infrastructures, consent approaches, governance), the public should be included in the vetting process. Initial efforts should create opportunities for inviting feedback on the acceptability of proposed models of research use. In addition, ongoing efforts will be needed to raise public awareness of what uses are made of their personal information for health research and what options are available for individuals to control the use of that information.
Beyond consulting with and informing the general public on research uses of their health information, serious consideration should be given to designing into the overall governance structure meaningful ongoing lay input into decision making and the introduction of benefit-sharing rules that allow for some financial benefit back to the community (e.g. toward further research). This could help with dealing with such thorny issues as commercialization and intellectual property in research outputs. A similar involvement of the public in governance has been suggested in the context of biobanks and stem cell research. (Haddow, Laurie, Cunningham-Burley & Hunter, 2007; Winickoff, 2006; Winickoff & Winickoff, 2003)
RECOMMENDATIONS:
- Operational models of use of personal health information for research (infrastructures, consent approaches, governance) should be vetted with all affected parties, including the public, in a variety of forums. Initial efforts should create opportunities for inviting feedback on the acceptability of proposed models of research use.
- Ongoing efforts will be needed to raise public awareness of what uses are made of their personal information for health research and what options are available for individuals to control the use of that information.
- Design meaningful ongoing lay input in the overall governance structure.
4. Conclusions
In this analysis, observational health research and privacy protection have both been framed as public goods. Areas of tension between the two have been identified as well as ways in which they may successfully co-exist. Changes at the margin are not sufficient. Several technical developments are coming together to signal an opportunity for fundamental changes in our approach to both observational research and to protection of confidentiality and security. These include the advent of the interoperable EHR, the development of research platforms through registries and biobanks, and the more intensive research use of medical record data as translational bioinformatics research comes on stream. Attention to the matter of necessary governance and infrastructures needs to be given sooner rather than later, lest the current “policy by procrastination” (Kosseim & Brady, 2008) lead to further insufficient and inefficient ad hoc amendments to the governance over research, rather than a holistic solution.
REFERENCES
Abbott,G. (2008). E-Health (Personal Health Information Access and Protection of Privacy) Act. Available at: http://www.leg.bc.ca/38th4th/3rd_read/gov24-3.htm.
Adida,B., & Kohane,I.S. (2006). GenePING: secure, scalable management of personal genomic data. BMC Genomics, 7, 93.
Advisory Council on Health Infostructure (1999). Canada health infoway. Paths to better health. Ottawa: Health Canada Publications.
Alberta Research Ethics Community Consensus Initiative (2005). ARECCI Recommendations - FINAL. Protecting People While Increasing Knowledge: Recommendations for a Province-wide approach to Ethics Review of Knowledge-generating Projects (Research, Program Evaluation, and Quality Improvement) in Health Care. Edmonton, AB: Alberta Heritage Foundation for Medical Research.
American Medical Informatics Association (2007). AMIA Strategic Plan. Available at: http://www.amia.org/inside/stratplan/.
Anonymous (1994). Report on statistical disclosure limitation methodology. Statistical policy working paper 22 (May 1994). Washington, DC: Statistical Policy Office, Office of Information and Regulatory Affairs, Office of Management and Budget.
Anonymous (2007a). UKCRC R&D Advisory Group to Connecting For Health: The Report of Research Simulations. UK Clinical Research Collaboration. Available at: http://www.ukcrc.org/pdf/CfH report June 07 full.pdf.
Anonymous (2007b). White Paper on Information Governance of the Interoperable Electronic Health Record (EHR). Ottawa: Canada Health Infoway Inc. Available at: http://www.infoway-inforoute.ca/Admin/Upload/Dev/Document/Information%20Governance%20Paper%20Final_20070328_EN.pdf.
Anonymous (2008a). Ensuring the inclusion of clinical research in the nationwide health information network. Available at: http://www.fastercures.org/pdf/FC_AHRQ-NCRR_report.pdf.
Anonymous (2008b). National Information Governance Board for Health and Social Care. Available at: http://www.connectingforhealth.nhs.uk/nigb.
Anonymous (2008c). NHGRI Seeks DNA Sequencing Technologies Fit for Routine Laboratory and Medical Use. Available at: http://www.genome.gov/27527585.
Anonymous (2008d). NIH Announces New Initiative in Epigenomics. Available at: http://www.nih.gov/news/health/jan2008/od-22.htm.
Black.C, McGrail,K., Fooks,C., Baranek,P., & Maslove,L. (2005). Data, Data, everywhere...: Improving access to population health and health services research data in Canada. Ottawa, Canada: Canadian Policy Research Networks.
Boettiger,S., & Bennett,A.B. (2006). Bayh-Dole: if we knew then what we know now. Nature Biotechnology, 24(3), 320-323.
Canada Health Infoway (2006). EHRS Blueprint Available at: http://knowledge.infoway-inforoute.ca/en/knowledge-centre/ehrs-blueprintv2.aspx.
Canadian Institutes of Health Research (2005). CIHR's Commercialization and Innovation Strategy. Available at: http://www.cihr-irsc.gc.ca/e/30162.html#2_Defining.
Canadian Institutes of Health Research (2008). CIHR Policy on Access to Research Outputs. Available at: http://www.cihr-irsc.gc.ca/e/32005.html.
Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, & Social Sciences and Humanities Research Council of Canada (2005). Tri-Council policy statement: ethical conduct for research involving humans, 1998 (with 2000, 2002, 2005 amendments). Ottawa: The Councils.
Canadian Institutes of Health Research Privacy Advisory Committee (2005). CIHR Best Practices for Protecting Privacy in Health Research - September 2005. Ottawa: Public Works and Government Services Canada. Available at: http://www.cihr-irsc.gc.ca/e/documents/pbp_sept2005_e.pdf.
Canadian Standards Association (1996). CAN/CSA-Q830-96, model code for the protection of personal information. A national standard of Canada. Ontario: Canadian Standards Association.
Caulfield,T., McGuire,A.L., Cho,M., Buchanan,J.A., Burgess,M.M., Danilczyk,U., Diaz,C.M., Fryer-Edwards,K., Green,S.K., Hodosh,M.A., Juengst,E.T., Kaye,J., Kedes,L., Knoppers,B.M., Lemmens,T., Meslin,E.M., Murphy,J., Nussbaum,R.L., Otlowski,M., Pullman,D., Ray,P.N., Sugarman,J., & Timmons,M. (2008). Research ethics recommendations for whole-genome research: consensus statement. Plos Biology, 6(3), e73.
Caulfield,T., Upshur,R.E.G., & Daar,A. (2003). DNA databanks and consent: A suggested policy option involving an authorization model. BMC Medical Ethics, 4 1-4.
Cavoukian,A. (2007). Order H0-004. Available at: http://www.ipc.on.ca/images/Findings/up-3ho_004.pdf. Ottawa: Information and Privacy Commissioner of Ontario.
Chamberlayne,R., Green,B., Barer,M.L., Hertzman,C., Lawrence,W.J., & Sheps,S.B. (1998). Creating a population-based linked health database: a new resource for health services research. Can J Public Health, 89(4), 270-273.
CIHR working group on case studies (2002). Secondary use of personal information in health research: case studies. Ottawa: Public Works and Government Service Canada. Available at: http://www.cihr-irsc.gc.ca/e/pdf_15568.htm.
Classen,J.B. (2006). Enhanced funding of pharmacoepidemiology through patenting the disclosure of adverse event information. Pharmacoepidemiology & Drug Safety, 15(6), 390-393.
Clayton,E.W., & Ross,L.F. (2006). Implications of disclosing individual results of clinical research. JAMA, 295(1), 37-38.
Coffey,M.J., & Ross,L. (2004). Human subject protections in genetic research. Genetic Testing, 8(2), 209-213.
Couzin,J. (2008). Genetic privacy. Whole-genome data not anonymous, challenging assumptions. Science, 321(5894), 1278.
Damschroder,L.J., Pritts,J.L., Neblo,M.A., Kalarickal,R.J., Creswell,J.W., & Hayward,R.A. (2007). Patients, privacy and trust: patients' willingness to allow researchers to access their medical records. Soc.Sci.Med., 64(1), 223-235.
Drolet,B.C., & Johnson,K.B. (2008). Categorizing the world of registries. Journal of Biomedical Informatics, 41(6), 1009-1020.
Eisenberg,R.S., & Nelson,R.R. (2002). Public vs. proprietary science: a fruitful tension?. Academic Medicine, 77(12:Pt 2), t-9.
El Emam,K. (2008). De-identifying health data for secondary use: A framework. Available at: http://www.ehealthinformation.ca/documents/SecondaryUseFW.pdf.
El Emam,K., Jonker,E., Sams,S., Neri,E., Neisa,N., Gao,T., & Chowdhury,S. (2007). Pan-Canadian De-identification Guidelines for Personal Health Information. Ottawa, Ontario. Available at http://www.ehealthinformation.ca/documents/OPCReportv11.pdf.
Eurostat (1996). Manual of disclosure control methods. Luxembourg: Office for Official Publications of the European Communities.
Fransen,G.A., van Marrewijk,C.J., Mujakovic,S., Muris,J.W., Laheij,R.J., Numans,M.E., de Wit,N.J., Samsom,M., Jansen,J.B., & Knottnerus,J.A. (2007). Pragmatic trials in primary care. Methodological challenges and solutions demonstrated by the DIAMOND-study. BMC Medical Research Methodology, 7, 16.
Freeman,P., & Robbins,A. (1999). The U.S. health data privacy debate. Journal of Technology Assessment in Health Care, 15(2), 316-331.
Gibson,E., Willison,D.J., Brazil,K., Coughlin,M.D., Emerson,C., Fournier,F., Schwartz,L., Szala-Meneok,K.V., & Weisbaum,K. (2008). Who's Minding the Shop? The Role of Canadian Research Ethics Boards in the Creation and Uses of Registries and Biobanks. BMC Med.Ethics, 9(17), doi:10.1186/1472-6939-9-17.
Grady,D.G., & Hearst,N. (2007). Using Existing Databases. In S.B. Hulley, S.R. Cummings, W.S. Browner, D.G. Grady, & T.B. Newman (Eds.), Designing Clinical Research (pp.207-221). Philadelphia: Wolters Kluwer / Lippincott Williams & Wilkins.
Grant,R.W., Wald,J.S., Poon,E.G., Schnipper,J.L., Gandhi,T.K., Volk,L.A., & Middleton,B. (2006). Design and implementation of a web-based patient portal linked to an ambulatory care electronic health record: patient gateway for diabetes collaborative care. Diabetes Technology & Therapeutics, 8(5), 576-586.
Haddow,G., Laurie,G., Cunningham-Burley,S., & Hunter,K.G. (2007). Tackling community concerns about commercialisation and genetic research: a modest interdisciplinary proposal. Social Science & Medicine, 64(2), 272-282.
Hess,R., Bryce,C.L., Paone,S., Fischer,G., McTigue,K.M., Olshansky,E., Zickmund,S., Fitzgerald,K., & Siminerio,L. (2007). Exploring challenges and potentials of personal health records in diabetes self-management: implementation and initial assessment. Telemedicine Journal & E-Health, 13(5), 509-517.
Iyengar,S.S., & Lepper,M.R. (2000). When choice is demotivating: can one desire too much of a good thing? Journal of Personality & Social Psychology, 79(6), 995-1006.
Jabine,T.B. (1993). Procedure for restricted data access. Journal of Official Statistics, 9 537-589.
Kennedy,D. (2005). Bayh-Dole: almost 25. Science, 307(5714), 1375.
Kho,M., Duffett,M., Willison,D.J., Cook,D.J., & Browers,M.C. (2008). Does written informed consent introduce selection bias in observational studies using medical records? A systematic review. BMJ, (Accepted for publication).
Kohane,I.S., Mandl,K.D., Taylor,P.L., Holm,I.A., Nigrin,D.J., & Kunkel,L.M. (2007). Medicine. Reestablishing the researcher-patient compact. Science.316(5826):836-7.
Kosseim,P., & Brady,M. (2008). Policy by procrastination: Secondary use of electronic health records for health research purposes. McGill Journal of Law and Health, 2 5-45.
Maclure,M., Carleton,B., & Schneeweiss,S. (2007). Designed delays versus rigorous pragmatic trials: lower carat gold standards can produce relevant drug evaluations. Medical Care, 45(10:Supl 2), Supl-9.
Malin,B., & Sweeney,L. (2001). Re-identification of DNA through an automated linkage process. Proceedings / AMIA, Annual Symposium-7.
Manson,N.C., & O'Neill,O. (2007). Rethinking informed consent in bioethics. New York: Cambridge University Press.
McGuire,A.L., & Gibbs,R.A. (2006). Genetics. No longer de-identified. Science, 312(5772), 370-371.
Medical Research Council (2008). Principles for access to, and use of, MRC funded research data. Available at: http://www.mrc.ac.uk/PolicyGuidance/EthicsAndGovernance/DataAccess/index.htm.
Meystre,S.M., Savova,G.K., Kipper-Schuler,K.C., & Hurdle,J.F. (2008). Extracting information from textual documents in the electronic health record: a review of recent research. Yearbook of Medical Informatics 128-144.
Miller,F.A., Giacomini,M., Ahern,C., Robert,J.S., & de,L.S. (2008). When research seems like clinical care: a qualitative study of the communication of individual cancer genetic research results. BMC Medical Ethics, 9 4.
Miller,F.G., & Pearson,S.D. (2008). Coverage with evidence development: ethical issues and policy implications. Medical Care, 46(7), 746-751.
Monane,M., & Avorn,J. (1996). Medications and falls. Causation, correlation, and prevention. Clinics in Geriatric Medicine, 12(4), 847-858.
National Information Governance Board (2007). The Care Record Guarantee. Our guarantee for NHS care records in England. Available at: http://www.connectingforhealth.nhs.uk/nigb/crsguarantee.
National Institutes of Health (2008). National Institutes of Health Public Access Policy. Available at: http://publicaccess.nih.gov/.
National Research Council (1997). For the Record. Protecting electronic health information. Washington: National Academy Press.
O'Neill,O. (2001). Informed consent and genetic information. Stud.Hist.Phil.Biol.& Biomed.Sci., 32(4), 689-704.
Ozimek,J. (2008). French storm the bastille over 'Sarkozy's Big Sister' database. Available at: http://www.theregister.co.uk/2008/09/11/france_database_tumulte/.
Pearson,S.D., Miller,F.G., & Emanuel,E.J. (2006). Medicare's requirement for research participation as a condition of coverage: is it ethical? JAMA, 296(8), 988-991.
Ram,N. (2008). Tiered Consent and the Tyranny of Choice. Jurimetrics, 48(3). Available at http://ssrn.com/abstract=1112364.
Robling,M.R., Hood,K., Houston,H., Pill,R., Fay,J., & Evans,H.M. (2004). Public attitudes towards the use of primary care patient record data in medical research without consent: a qualitative study. J.Med.Ethics, 30(1), 104-109.
Rothstein,M.A., & Talbott,M.K. (2006). Compelled Disclosure of Health Information: Protecting Against the Greatest Potential Threat to Privacy. JAMA: The Journal of the American Medical Association, 295(24), 2882-2885.
Sanders,D., & Protti,D. (2008). Data Warehouses in Healthcare: Fundamental Principles. Electronic Healthcare, 6(3), 1-16. Available at: http://www.longwoods.com/product.php?productid=19510&cat=524&page=1.
Shaffer,C. (2007). Next-generation sequencing outpaces expectations. Nature Biotechnology, 25(2), 149.
Singleton,P., & Wadsworth,M. (2006). Consent for the use of personal medical data in research. BMJ, 333(7561), 255-258.
Slaughter,P.M., Collins,P.K., Roos,N., Weisbaum,K.M., Hirtle,M., Williams,J., Martens,P.J., & Laupacis,A. (2006). Harmonizing research & privacy: Standards for a collaborative future. Privacy best practices for secondary data use (SDU). [CD-ROM].: Institute for Clinical Evaluative Sciences and Manitoba Centre for Health Policy.
Subgroup on Procedural Issues for the TCPS (ProGroup) (2008). Proposed Textual Changes to REB Operational Issues in the Tri-Council Policy Statement: Ethical Conduct for Research Involving Humans (TCPS). Ottawa: Interagency Panel on Research Ethics. Available at: http://www.pre.ethics.gc.ca/english/workgroups/progroup/REB_Operational_Issues.cfm.
Tezeta,F.M.a.K. (2008). ICES Report: Using Data from Electronic Medical Records: Theory versus Practice. Healthcare Quarterly, 11(4), 23-25.
The Experts Committee for Human Research Participant Protection in Canada (2008). Moving Ahead: Final Report. Available at: http://www.hrppc-pphrc.ca/english/sponsors.html. Ottawa.
Thursby,J.G., & Thursby,M.C. (2003). Intellectual property. University licensing and the Bayh-Dole Act. Science, 301(5636), 1052.
Walker,A.M. (2006). More lawyers. More bureaucrats. Less information on drug safety. Pharmacoepidemiology & Drug Safety, 15(6), 394-395.
Westfall,J.M., Mold,J., & Fagnan,L. (2007). Practice-based research--"Blue Highways" on the NIH roadmap. JAMA, 297(4), 403-406.
Willison,D.J., Emerson,C., Szala-Meneok,K.V., Gibson,E., Schwartz,L., Weisbaum,K., Fouriner,F., Brazil,K., & Coughlin,M.D. (2008a). Access to medical records for research purposes: Varying perceptions across Research Ethics Boards. J Med Ethics, 34 308-314.
Willison,D.J., Kapral,M.K., Peladeau,P., Richards,J.A., Fang,J., & Silver,F.L. (2006). Variation in recruitment across sites in a consent-based clinical data registry: lessons from the Canadian Stroke Network. BMC Medical Ethics, 7(6), doi:10.1186/1472-6939-7-6.
Willison,D.J., Schwartz,L., Abelson,J., Charles,C., Swinton,M., Northrup,D., & Thabane,L. (2007). Alternatives to project-specific consent for access to personal information for health research: What is the opinion of the Canadian public? Journal of the American Medical Informatics Association, 14 706-712.
Willison,D.J., Swinton,M., Schwartz,L., Abelson,J., Charles,C., Northrup,D., Cheng,J., & Thabane,L. (2008b). Alternatives to project-specific consent for access to personal information for health research: insights from a public dialogue. BMC Med.Ethics, 9(18), doi:10.1186/1472-6939-9-18.
Winickoff,D.E. (2006). Governing stem cell research in California and the USA: towards a social infrastructure. Trends in Biotechnology, 24(9), 390-394.
Winickoff,D.E., & Winickoff,R.N. (2003). The charitable trust as a model for genomic biobanks. New England Journal of Medicine, 349(12), 1180-1184.
Wolf,L.E., Zandecki,J., & Lo,B. (2004). The certificate of confidentiality application: a view from the NIH Institutes. Irb: a Review of Human Subjects Research, 26(1), 14-18.
Wolf,S.M., Lawrenz,F.P., Nelson,C.A., Kahn,J.P., Cho,M.K., Clayton,E.W., Fletcher,J.G., Georgieff,M.K., Hammerschmidt,D., Hudson,K., Illes,J., Kapur,V., Keane,M.A., Koenig,B.A., Leroy,B.S., McFarland,E.G., Paradise,J., Parker,L.S., Terry,S.F., Van,N.B., & Wilfond,B.S. (2008). Managing incidental findings in human subjects research: analysis and recommendations. Journal of Law, Medicine & Ethics, 36(2), 219-248.
Yeates,N., Lee,D.K., & Maher,M. (2007). Health Canada's Progressive Licensing Framework. CMAJ Canadian Medical Association Journal, 176(13), 1845-1847.
Alternate versions
- PDF (193 KB) Not tested for accessibility
- Date modified: